祖先重组图的推断与应用-文献精读59
Inference and applications of ancestral recombination graphs
祖先重组图的推断与应用

摘要
祖先重组图(ARGs)总结了在一组DNA序列样本中个体之间复杂的系谱关系。ARGs的应用正在推动群体遗传学领域的革命,促使强大新方法的发展,用于阐明个体和群体的遗传过程,包括群体大小历史、迁移、杂合、重组、突变和选择。在本综述中,我们向读者介绍了ARGs的结构,并讨论它们与重组和遗传漂变等过程的关系。我们还探讨了估算ARGs方法的异同,并提供了具体的实例,说明ARGs如何用于揭示群体层面的过程。
引言
自PCR和DNA测序技术发明以来,合并理论一直是群体遗传数据分析的基础。合并理论的核心对象是合并树(图1),它总结了样本中个体之间的遗传关系信息。树的叶(末端)代表样本中的个体DNA序列;边(也称为谱系或分支)代表遗传下传的血缘线;内部节点代表合并事件(也称为合并事件),即样本中特定个体或个体组的谱系在时间点上合并(或“合并”)到他们最近的共同祖先(MRCA)。树的根代表样本中所有个体的最近共同祖先。
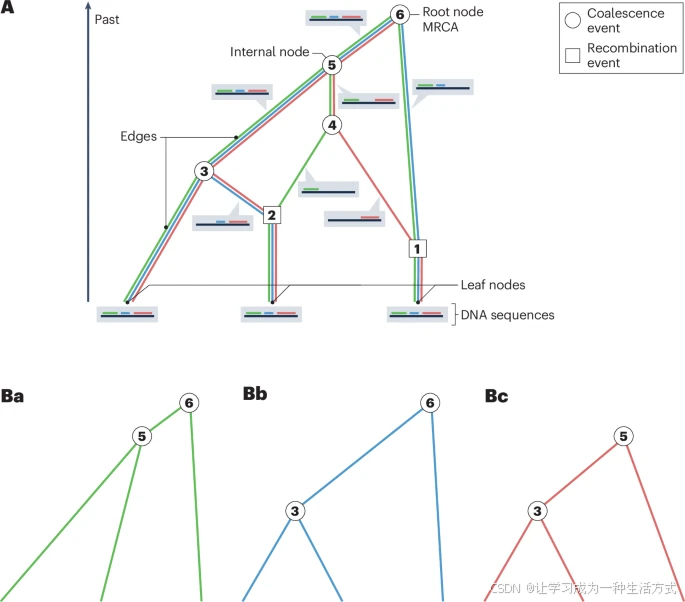
A,三个样本DNA序列的祖先重组图(ARG)示例,追溯合并过程,直到找到最近的共同祖先(MRCA)。合并树由一组由边连接的节点组成。代表观察到的DNA序列的节点是树中的叶(叶节点),代表合并事件的节点是内部节点。树顶端的节点代表这三个序列的MRCA。在该图中,仅对内部节点编号,并以圆圈表示叶节点。此外,树的绘制方式是根在顶部,叶在底部,这是计算机科学中的传统。在ARG中,圆圈代表合并事件,方形代表重组事件。谱系上的每个重组事件将该谱系代表的DNA序列分割为两个片段,因此这些样本的历史中的两个重组事件(1和2)将样本序列分割为三个片段(分别标记为绿色、蓝色和红色),每个片段都有一个独特的合并树。合并事件(3, 4, 5和6)将谱系合并到一个共同祖先中。请注意,谱系代表包含基因组某些片段(或多个片段)遗传材料的血统,这些片段是样本中一个或多个个体的祖先遗传材料。包含三个DNA序列祖先遗传材料的DNA片段相邻于每个边显示。例如,从节点2到节点4的谱系仅包含DNA序列中第一个绿色片段的祖先材料。该谱系上的其余DNA序列并不是样本中三位个体的祖先材料。
B,三个DNA序列片段(绿色、蓝色和红色)的合并树。所有三个合并树都嵌入在ARG中(A部分),即可以从ARG推导出。尽管拓扑结构相同,但这些树因分支长度不同而有所区别。
金曼(Kingman)和哈德森(Hudson)在20世纪80年代早期发现,群体样本中的个体关系可以用二叉树来描述,即每个节点要么没有子节点(叶节点),要么有两个子节点(内部节点),而群体遗传学模型中的遗传漂变能够对树的结构作出简单预测。这一发现很快促使许多统计方法的出现,用于推断诸如群体大小变化和迁移率等群体级别的参数,并推动了利用群体遗传数据检测自然选择的新方法的发展。这些方法的核心洞见是:DNA测序数据中关于群体级过程的所有信息都体现在合并树中。如果合并树能够以100%的准确率推断出来,那么从序列数据中关于这些人口学过程的信息将不再有所增加。这一原则推动了全似然和贝叶斯统计方法的发展,这些方法通过使用马尔可夫链蒙特卡洛(MCMC)或其他随机方法对可能的合并树集进行积分,实现了其目标;换句话说,它们通过考虑许多树并依据相对概率对其加权,来处理合并树结构的不确定性。
到2003年人类基因组被测序时,已经形成了一套成熟的基于树的群体遗传数据分析的统计方法。然而,唯一的问题是:这些方法假设没有重组,因此只适用于线粒体DNA、叶绿体DNA、Y染色体、一些病毒测序数据以及其他一些不受重组影响的标记。然而,随着新一代测序技术的出现,这些方法迅速过时了,因为重点转移到包含大量重组的基因组数据上。对于基因组数据,并不只有一棵合并树,而是存在与数据历史中的每个重组事件对应的多棵合并树。每次重组事件会将DNA序列分成两个独立的片段,每个片段将有不同的合并过程,从而在重组断点的两侧形成不同的树。一些方法试图通过假设基因组可以分为短小的基因组区域,区域之间自由重组,但区域内部无重组来解决这一问题。然而,人类的突变率和重组率相对相似,这意味着平均而言,每次新突变发生时可能提供有关树结构的信息,而新的重组事件也会发生,改变树结构。此外,许多针对短小无重组区域的方法无法在计算上扩展到数千个基因组区域。因此,该领域逐渐远离分析合并树的全似然和贝叶斯方法,转而使用复合似然方法,假设SNPs是独立的,分析等位基因频率分布,使用近似贝叶斯计算,重点分析序列对或其他仅利用数据中的一小部分信息的方法。这是该领域的现状,直到2014年,研究表明在带有重组的模型中推断合并树实际上在计算上是可行的,即使对于基因组规模的数据也是如此。通过巧妙的采样方法,有可能沿基因组长度估计(并从后验分布中采样)联合结构,即祖先重组图(ARG)中的合并树。这一发展为从群体遗传测序数据中提取更多信息提供了可能,并可能开发出利用ARG替代单个合并树的全似然和贝叶斯方法用于群体遗传推断。
祖先重组图
ARGs 和带有重组的合并(CwR)
1983年首次描述的带有重组的合并过程(CwR)被构建为一个从现在向过去运行的随机过程,允许重组事件和合并事件发生,直到为每个位点找到最近的共同祖先(MRCA)。在后来的工作中,该过程得到了进一步的发展,并创造了"ARG"这个术语,用于描述由最初描述的CwR产生的随机图结构。ARGs 的基本概念是,它们代表了一个合并过程,其中祖先谱系不仅通过合并合并,还因重组而分裂。通过ARG,可以推导出基因组每个片段的个别合并树。重组通常会生成拓扑结构不同的树,尽管有时它们可能只是分支长度不同。ARG包含有关所有基因组合并树的信息,同时还包含有关重组事件及其在线系中的位置的信息。显然,对于包含许多个体和数万到数十万次重组事件的基因组数据,ARG可能会变得非常复杂。因此,通常会进行一些简化。特别是谱系上重组事件的时间无法从数据中推断出来,因此通常不会在ARG中表示。此外,通常还会使用进一步的简化和近似来使基于ARG的推断变得可行。
ARGs作为树的序列
生成ARGs的随机过程追踪从现在向过去的重组和合并事件。另一种看待生成ARGs过程的方式是考虑沿基因组长度合并树是如何变化的。因为可以从ARG推导出树,所以我们可以考虑一个从染色体一端到另一端运行的过程,生成考虑到影响每个基因组片段的特定重组和合并事件的合并树。该过程被证明是非马尔可夫的,这意味着某个位置的合并树的概率分布不仅依赖于前一个位置的合并树,还依赖于染色体长度上的所有其他树。因此,考虑沿染色体长度变化的树的序列过程在计算上相当复杂,因为需要同时跟踪所有树以进行推断和模拟等目的。
为了获得更简单且在计算上更可处理的过程,提出了一种模型,可以将沿基因组长度的树生成过程近似为马尔可夫过程,称为顺序马尔可夫合并(SMC)过程。该想法是只允许那些祖先遗传材料重叠的谱系之间发生合并事件。例如,在图1A中,两个谱系的合并事件分别仅包含绿色和红色基因组片段的祖先遗传材料,形成了节点4。在SMC过程中,不允许这样的合并事件,因为在特定位置的合并树只能依赖于前一个位置的树,从而简化了过程。
虽然SMC过程只允许那些包含至少一些共享祖先遗传材料的谱系之间的合并事件,但SMC过程的一个改进版本,称为SMC′,还允许那些没有共享遗传材料但在相邻基因座处含有遗传材料的谱系之间发生合并事件。这种SMC的小扩展不会增加太多的计算复杂性,但它极大地提高了近似的准确性。后续的研究表明,SMC′过程在大多数方面都是完整CwR的非常接近的近似,并且今天可能是用于群体遗传推断的首选CwR近似模型。CwR、SMC或SMC′模型下允许的合并事件在图2中进行了详细说明。
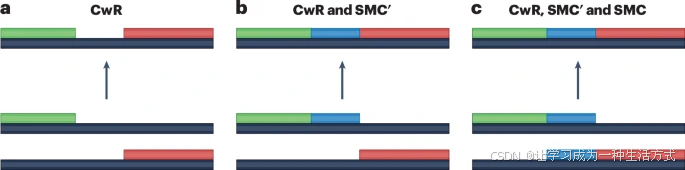
a. 两条携带不重叠且非相邻DNA片段的谱系之间的合并事件。此类合并事件仅在完整的带有重组的合并模型(CwR)下允许,而在SMC′或SMC模型中不允许。 b. 两条携带相邻但不重叠DNA片段的谱系之间的合并事件。此类合并事件在CwR和SMC′模型下允许,但在SMC模型中不允许。 c. 重叠DNA片段之间的合并事件。此类合并事件在所有三种模型中都允许。SMC′和SMC模型对允许的合并类型的限制,缩小了在这些模型下可以模拟的祖先重组图(ARG)的状态空间,其中SMC施加的限制比SMC′更为严格。SMC指的是顺序马尔可夫合并模型。
关于生成ARGs过程的不同假设导致了ARGs的不同定义。一些在完整CwR模型中允许的ARGs不被SMC′模型支持,而一些被SMC′模型支持的ARGs在SMC模型中不被允许。有时,ARGs可以被更简单地表示为一系列树,不包含关于重组的明确信息,或者不标识哪些合并事件在树之间共享。在这种情况下,这些树的序列是否是真正的ARGs就成为一个定义问题。然而,无论ARGs如何定义,它们都可以被视为由群体遗传因素(如群体大小、迁移率和选择)以及分子力量(如突变和重组)控制的合并过程的产物。ARGs包含关于这些过程的信息,基于ARGs的方法能够提取这些信息,有潜力在基因组时代群体遗传学中扮演核心角色,类似于1990年代合并树的角色。
ARGs的推断
ARGs的表示与推断范式
已经开发了许多推断ARGs的方法,包括Margarita、ARGweaver、RentPlus、Arbores、Relate、tsinfer + tsdate、ARGweaver-D、SARGE、KwARG、ARGinfer、ARG-Needle和SINGER等。我们注意到,tsinfer和tsdate是分别开发的工具,分别用于推断ARG的拓扑结构和内部节点的年龄;我们用“tsinfer + tsdate”指代这两种方法的联合使用。这些方法在许多方面有所不同,包括它们表示ARGs的方式,这与关于潜在生成过程的隐含假设有关。Relate和RentPlus等方法将ARGs表示为一系列合并树,并不强制要求这些树共享节点或分支。这种表示方式有时是方便的,但效率不高,因为嵌入ARGs的许多树共享相同的节点和边,尤其是在样本量大的情况下,这在存储上会变得非常低效。此外,这种表示方式破坏了一些自然发生的树之间的相关结构。相关树序列的概念是模拟算法(如FastCoal、fastsimcoal/fastsimcoal2和ms)以及推断程序(如tsinfer、ARG-Needle、SINGER和ARGweaver)的基础。它还可以导致紧凑的“简洁树序列”数据结构,用于序列数据,如在广泛使用的模拟软件msprime中实现的那样,在其中每个节点和边只需要表示一次,即使它可能是许多树的组成部分。将ARG表示为一组共享节点和边的树序列的这一表示方式显著提高了模拟速度和存储效率,许多当前的ARG推断方法提供了以这种紧凑格式生成输出的选项(例如,tsinfer、ARG-Needle和SINGER)。msprime引入了这一数据格式,是最常用的ARGs模拟软件,也是第一个充分利用这种数据结构进行快速ARG模拟的软件。
ARGs的推断是一个计算上非常困难的问题,因为可能的图拓扑数量可能是巨大的。此外,观察到的序列数据提供了关于基因组特定位置局部树结构的有限信息。一些方法,如ARGweaver/ARGweaver-D、ARGinfer和SINGER,通过采用贝叶斯方法解决了这一问题,在该方法中,ARGs是通过MCMC从后验分布中采样的。与其推断单个ARG,不如采样多个可能的ARG,这提供了ARG推断中的不确定性表示,并有助于下游的严格统计分析。尽管Relate和tsinfer + tsdate推断了一个固定的ARG拓扑结构,Relate还采样了推断拓扑结构的合并时间,而tsdate输出了关于每个节点年龄的边缘分布的元数据。大多数其他方法目前仅提供ARG拓扑结构和分支长度的点估计。由于ARGs的采样在计算上要求很高,估计单个ARG的方法往往比采样多个ARG的方法快得多。
可扩展性、准确性、数据要求与输出
ARG推断方法正在快速发展,不断改进。我们在此提供一个概述,并引用了最近关于各种方法的综合比较,这些比较衡量了进化推断的各个方面,例如重组、合并时间、等位基因频率和多基因分数历史。
当前的ARG推断方法在模型假设、计算速度、推断准确性、进行推断的类型以及支持的数据类型等方面有所不同。例如,一些方法需要经过相位的现代数据,而其他方法可以使用其他类型的数据,包括古代DNA和基因分型阵列数据。ARGweaver和ARGweaver-D是唯一可以使用未经相位数据的方法;tsinfer-sparse(tsinfer的一种变体)和ARG-Needle是唯一支持基因分型阵列数据的方法。ARGweaver-D、Relate、tsinfer + tsdate 和 SARGE可以支持古代DNA的使用,除了ARGweaver-D外,所有程序都需要对古代DNA进行计算填补并相位。此外,Relate和tsinfer还要求对祖先状态进行极性化,通常使用外群物种推断祖先状态。不同方法的适用性在表1中进行了总结。
| Samples branch lengths | Samples topologies | Scales to 1000 Genomes Project | Scales to UK Biobank | Supports ancient DNA | Supports genotyping array | |
|---|---|---|---|---|---|---|
| ARGweaver | Yes | Yes | No | No | Yes | No |
| RentPlus | No | No | No | No | No | No |
| Arbores | Yes | Yes | No | No | No | No |
| Relate | Yes | No | Yes | No | Yes | No |
| tsinfer+tsdate | No | No | Yes | Yes | Yes | Yes |
| ARGweaver-D | Yes | Yes | No | No | Yes | No |
| SARGE | No | No | No | No | No | No |
| KwARG | No | No | No | No | No | No |
| ARGinfer | Yes | Yes | No | No | No | No |
| ARG-Needle | No | No | Yes | Yes | No | Yes |
| SINGER | Yes | Yes | No | No | No | No |
-
The wording ‘Samples branch lengths’ and ‘Samples topologies’ refers to whether the methods just provide a single point estimate or sample multiple instances to provide measures of statistical uncertainty. ARG, ancestral recombination graph.
在计算速度方面,像ARGweaver、Arbores、KwARG和ARGinfer等方法只能处理几十条序列,其中KwARG和ARGinfer也仅限于处理相对较短的序列。RentPlus、SARGE和SINGER等方法可以处理数百条全基因组序列,Relate可以处理数千条序列,包括完整的1000基因组计划数据。当前最具可扩展性的方法是tsinfer和ARG-Needle,它们能够处理数十万条序列,包括完整的UK Biobank数据以及其他大型全基因组关联研究(GWAS)数据。
模拟研究发现,最快的方法通常(但不总是)表现最差,这并不令人意外,因为在ARG推断问题中,与许多其他计算问题一样,计算速度与准确性之间存在权衡。然而,具有相似速度的方法有时在统计性能上差异很大,通常更新的方法表现优于较老的方法。我们建议读者参考最近的模拟比较,以了解各种方法的准确性细节。
正如之前讨论的那样,这些方法在是否提供统计不确定性的测量上也有所不同,其中ARGinfer、ARGweaver、ARGweaver-D和SINGER是唯一提供统计不确定性全概率建模的方法。最后,这些方法在提供的ARG表示形式上也有所不同,Relate等方法在窗口中估计树,而SINGER、ARGweaver和ARGweaver-D等其他方法则提供表示一系列树、并由个别重组事件分隔的图。
ARGs在群体遗传推断中的应用
人口统计学和选择的推断
ARGs可以用于对群体遗传过程进行详细而有力的推断,例如群体大小历史、迁移、自然选择、突变率和重组率。ARGs的一个主要应用领域是推断人口统计学和个体之间的系谱关系。某个时间点的合并速率与该时刻的有效种群大小(Ne)成反比。因此,可以通过ARG的合并时间的时间密度来估计Ne随时间的变化(图3a)。
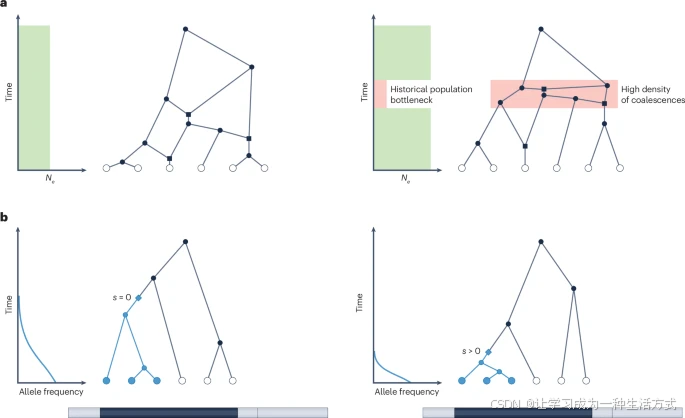
a. 历史人口规模的变化反映在祖先重组图(ARG)中。左侧图显示了在人口规模恒定的情况下的ARG。右侧图显示了在历史人口瓶颈中的ARG。在这一时期,较小的有效种群大小(Ne的减少)导致整个ARG中的合并事件密度高于人口规模恒定的历史。方形代表重组事件,圆形代表合并事件。 b. 在一个SNP上的自然选择会导致被选择等位基因的频率迅速增加,从而导致携带该等位基因的谱系在SNP所在的树中的合并密度增加(用蓝色表示)。两个面板均显示了基因组特定区域的合并树(标记为深灰色);需要注意的是,并未显示整个ARG。某条谱系上的突变(显示为蓝色菱形)导致所有后代谱系继承该等位基因。如果该等位基因是中性的(s=0)(左侧),预计不会有合并密度的差异。然而,如果该等位基因是有利的(s>0)(右侧),则预计蓝色谱系的合并密度会增加。
ARGs还被用于通过提取整个基因组的合并树并考虑每棵树中谱系的不同可能迁移历史,来估计复杂人口统计模型的参数。这种考虑谱系通过人口统计模型的可能路径的思想也被用于局部祖先推断,其中基因组中的每棵树提供了该树通过人口统计模型的路径信息,从而推断该树所覆盖片段的局部祖先信息。ARGs还可以用来推断自然选择。利用ARGs估计单个SNP的选择系数的推断方法,通过提取焦点SNP周围的局部树,然后检查携带衍生等位基因的谱系与携带祖先等位基因的谱系的相对合并速率。例如,一个正向选择的等位基因(s>0)预计在时间前进时比中性等位基因(s=0)的频率增加得更快。因此,向时间倒退时,正向选择的等位基因会显得其频率迅速下降,这意味着携带该等位基因的谱系数量会快速减少。我们预计这些携带选择等位基因的谱系的合并事件会有高密度,类似于之前讨论的小种群大小产生高密度合并事件的情况。通过分析携带不同等位基因的谱系的合并密度来推断选择,可以通过显式计算似然并对衍生等位基因频率进行积分来完成,也可以通过对推断出的ARG本身应用机器学习技术来完成。这些方法还可以稍作修改,以同时研究多个SNP上的选择作用,即多基因选择,或者通过时间重建多基因评分。
除了推断人口统计和选择外,ARGs还被用于推断的其他令人兴奋的应用,例如将推断出的ARGs与线性混合模型框架结合,改进复杂性状变异的关联分析。该方法的一个特别优势是在稀疏基因分型阵列数据上准确检测稀有变异的大效应关联。另一个有前景的研究领域是使用ARGs来显式建模给定样本的祖先谱系的空间位置。此外,ARGs还被用于通过检查突变映射到ARG分支上的方式,研究人类突变谱的演化,研究突变频率随时间的变化。类似的考虑突变可能映射到合并树分支的隐式方法被用于估算等位基因的年龄,进而用于推断不同人类群体中的历史世代时间。虽然关于具体使用方法存在争议,但这一框架依然展示了ARGs在帮助理解复杂的分子和人口统计过程中的令人兴奋的潜力。新兴的基于ARGs的方法也已被开发出来,用于推断同祖段、检测染色体倒位、计算分层的基于树的f统计量、模拟和分析定量性状、建模连锁不平衡以及模拟局部祖先。此外,将ARGs整合到前向模拟中,也极大扩展了ARGs可以应用的推断问题范围,因为前向模拟可以生成比时间倒退模拟更广泛的分子和群体水平过程。
使用ARGs进行完整的概率推断
给定数据集可能有许多ARGs,但使用ARGs的分析通常依赖于从数据中估计的单个ARG。将单个推断的图视为真实图极大地简化了后续分析,但这带来了两个问题。首先,这没有考虑基础ARG的不确定性,导致任何使用单个ARG作为真实ARG的分析结果中出现虚假的确定性。其次,任何ARG都是在某些隐式或显式假设下推断的(例如无选择、种群大小恒定、无种群结构),因此会有偏差,因为真实的群体遗传参数可能与假设情景不同。因此,ARG的未观察性需要对ARG的不确定性进行积分和偏差校正,以实现完整的概率推断。
未来的方法可以通过两种方式之一来利用ARGs进行完整的概率推断。第一种方式是通过一种称为重要性采样的统计技术。在这种版本的重要性采样中,ARGs是基于特定模型和特定参数假设(如有效种群大小或选择系数)采样的。然后,这些ARGs根据它们在采样模型下的概率被赋予权重,权重与其概率成反比。通过适当重新加权样本,可以近似任意参数值处的群体遗传参数的似然函数,从而提供参数估计。在最初用于后验采样的参数集下更可能的ARGs被赋予较低权重,而那些较不可能的ARGs则被赋予较高权重。最后,可以将采样的ARGs的概率加权平均来近似任何参数值的似然。随着使用的ARG样本量增加,这一近似将变得更加精确。
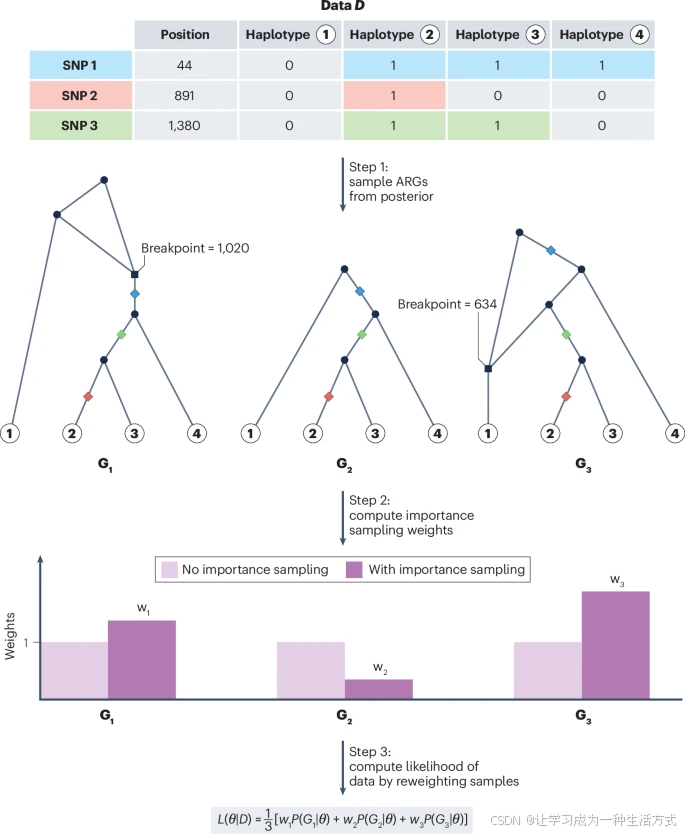
在第1步中,根据某个模型从特定的后验分布中采样出许多祖先重组图(ARGs)。为了简化展示,这里只显示了三个采样的图。与图3类似,突变在ARG中以彩色菱形表示,方形代表重组事件,圆形代表合并事件。在第2步中,计算每个图的重要性采样权重。在第3步中,我们通过每个采样图的加权平均似然值来计算参数的似然值。这里,D代表序列数据,(G1–G3) 代表不同的采样ARG,(w1–w3) 代表这些ARGs相应的重要性采样权重。θ代表任何感兴趣的群体遗传参数,比如种群大小或选择系数。
与重要性采样的替代方法是将参数估计直接整合到用于采样ARGs的MCMC算法中。例如,除了提议ARG拓扑结构和分支长度的变化外,ARG采样算法还可以提议历史种群大小或突变率的变化。之前已有一些工作在用户定义的人口统计模型下推断ARG,并且Relate具备在估计合并时间和历史种群大小之间交替进行的能力,但尚未有方法可以同时采样ARGs和参数。基于联合采样概念的框架已经被开发出来,但它们主要集中于小样本量或总重组事件较少的情况。这很可能是因为在执行联合采样时计算成本显著增加,同时还必须将每个感兴趣的参数整合到ARG推断算法中。相比之下,重要性采样可以在不改变基本推断算法的情况下进行,因此可能为推断更广泛的参数集提供更大的灵活性。例如,可以设想使用ARGs来研究其他过程,如背景选择、择偶交配、混血图的推断。因此,我们认为使用ARG的重要性采样进行群体遗传参数的完整概率推断是群体遗传学领域未来研究的重要方向。该领域的其他重要未来方向包括开发快速、适当校准的ARG推断方法,并找到高效计算ARG或合并树在给定群体遗传参数集下的概率的方法。
在人类数据上的应用示例
为了说明推断出的ARGs在理解群体遗传过程中的实用性,我们提供了三个例子。我们首先将SINGER应用于围绕ABO基因和MCM6基因的人类测序数据。两者都是先前报告的自然选择目标,但这里我们展示如何使用推断出的ARGs探索选择信号。
决定个体血型的ABO基因是HLA区域外变异性最大的编码区之一。比较基因组学分析显示,该位点存在跨物种多态性的证据,并且有假设认为维持这种多态性的平衡选择在人类和其他灵长类中起作用。如果这一假设成立,那么我们预计在ABO基因中会看到异常古老的最近共同祖先(TMRCA)。我们绘制了来自1000基因组计划CEU人群(北欧及西欧祖先的犹他州居民)中ABO基因位点周围1千碱基对的TMRCA估计值,并将其与第9号染色体的中位TMRCA(1.62百万年前)进行比较,结果显示该区域内的推断TMRCA超过了600万年前(大约是人类与黑猩猩的物种分化时间)。这些结果与平衡选择维持该位点的跨物种多态性假设相符。需要注意的是,由于SINGER使用了标准的合并先验,它可能低估了最古老的合并时间,这意味着该位点的真实TMRCA可能比这里显示的还要古老。

a. 紫色线条显示了从1000基因组计划中CEU人群(北欧及西欧祖先的犹他州居民)50条序列推断出的1千碱基对的最近共同祖先时间(TMRCA),绿色实线表示人类和黑猩猩的物种分化时间(约600万年前)。ABO基因(阴影区域)表现出比物种分化时间更古老的合并时间。 b. 祖先重组图(ARG)推断出的英国人群(GBR)所有样本(红色)与携带rs4988235衍生等位基因个体(蓝色)的1千碱基对分支长度多样性(基于1000基因组计划)。推断的ARG可在以下网址获取:ARG_review/inferred_ts at main · YunDeng98/ARG_review · GitHub。
rs4988235 SNP位点的衍生等位基因位于MCM6基因中,已被确定为乳糖持久性在欧洲人的因果变异,据信在乳制品农业引入后受到正向选择。将1000基因组计划中所有英国样本的整体1千碱基对分支长度多样性与携带rs4988235衍生等位基因单倍型的多样性进行比较,显示携带者内部的多样性明显低于整体多样性,并且携带者之间多样性的减少范围非常广。这一观察结果与先前提出的强烈、近期的选择性清扫一致。
我们还展示了ARGs推断不同人类群体人口历史的能力。我们使用tsinfer + tsdate对大量现代和古代基因组推断的ARG进行分析,考虑了四个群体:来自1000基因组计划的汉族(CHB)、英国人群(GBR)、约鲁巴人群(YRI),以及来自Simons基因组多样性计划的南美洲土著群体克丘亚人群。计算每个群体的全基因组内群体成对TMRCA的分布,显示约鲁巴人群具有最高密度的较早合并时间,因为非洲个体的祖先谱系未受到影响所有非洲外群体的“走出非洲”瓶颈的影响。相比之下,汉族和英国人群的祖先谱系受到该瓶颈的影响,表现出非常古老的成对TMRCA数量减少,同时更多的近期合并事件。克丘亚人群的TMRCA向近期极端倾斜,反映了克丘亚谱系既受到了“走出非洲”瓶颈的影响,也受到了在上一个冰川最大期期间,一部分人类迁徙通过白令陆桥定居美洲时所经历的瓶颈的影响。

a. 四个人群内成对最近共同祖先时间(TMRCA)的全基因组分布:来自1000基因组计划的约鲁巴、汉族、英国人群,以及来自Simons基因组多样性计划的克丘亚人群。对于每个人群,选择了三位个体,并在全基因组范围内每100千碱基对间隔计算这些个体谱系之间的成对TMRCA。这灵感来源于参考文献39,该文献比较了非洲和非洲外人群在同一个祖先重组图(ARG)中的TMRCA。推断出的ARG可在以下网址获取:A unified genealogy of modern and ancient genomes: Unified, inferred tree sequences of 1000 Genomes, Human Genome Diversity, and Simons Genome Diversity Projects。 b. 使用1000基因组计划推断的约鲁巴、汉族、英国和芬兰人群的历史有效种群大小。通过在第1号染色体上运行Relate获得。这灵感来源于参考文献35。
我们还在1000基因组计划的四个人群(汉族、英国、约鲁巴和芬兰人群)中运行了Relate,使用EstimatePopulationSize功能,该功能根据推断出的合并事件密度推断不同时期的历史种群大小,我们重建了每个人群的人口统计历史。我们观察到,在非常古老的时间里,所有人群似乎具有相同的种群大小,这符合这些人群尚未分化,仍属于同一祖先种群的预期。在1万至20万年前,所有非洲外人群的种群规模相对于约鲁巴人群显著减少,这与“走出非洲”瓶颈相吻合。在最近的时期,我们观察到芬兰人群的种群规模相对于英国人群减少,这与现代芬兰的已知小型创始种群相比于其他欧洲人群的情况一致。
结论
几年前,利用全基因组数据进行完整的概率群体遗传推断似乎是不可能的。然而,新的计算方法促进了ARGs的使用,解决了群体遗传学中的若干问题,例如推断突变年龄、识别个体间的精细关系、检测作用于性状和个体等位基因的自然选择。虽然仍处于初期阶段,但使用MCMC或重要性采样的ARG方法有望提供概率框架,充分利用全基因组数据中的丰富信息。即使没有这些方法,ARG推断仍然是可视化遗传变异和详细遗传关系的关键工具。并非所有ARG推断方法的表现都一样好。一般而言,准确性与计算复杂性之间存在权衡。此外,提供统计准确性衡量标准的方法(例如,ARGweaver和SINGER等全贝叶斯方法)通常比不提供统计不确定性衡量标准的方法慢得多。目前,只有ARG-Needle和tsinfer等方法适用于GWAS规模的数据集。我们预计在未来几年内,将会有许多进展改进ARG推断的计算和统计方面,并扩展ARG的应用,使群体遗传学完全进入基因组时代。